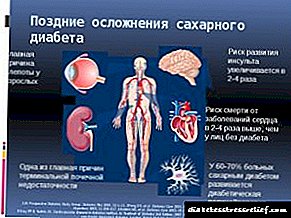
Ketonurija je prisustvo u urinu ketonskih tijela, naime acetona
Mehanizam ketonurije.
U zdrave osobe ugljikohidrati, lipidi i neki proteini oksidiraju do ugljičnog dioksida i vode. U nekim patološkim stanjima, posebno kod dijabetesa, smanjuje se proizvodnja inzulina. U jetri se smanjuju rezerve glikogena, mnoga tjelesna tkiva su u gladi energije. U tim se uvjetima aktiviraju procesi oksidacije proteina i lipida u jetri, ali nedostatak glikogena dovodi do njihove nepotpune oksidacije i nakupljanja u krvi neoksidiranih produkata metabolizma lipida i proteina - ketonskih tijela. Zbog nakupljanja ketonskih tijela u krvi (ketonemija), pH krvi se premješta u kiselu stranu. Ovo se stanje naziva acidoza. Urin takvog pacijenta ima oštru kiselinsku reakciju i mirise na aceton.
Glad, upotreba uglavnom proteinske i masne hrane, isključenje ugljikohidrata iz prehrane dovodi do povećanog stvaranja ketonskih tijela i njihovog izlučivanja mokraćom.
U ranoj dobi ketonurija je češća nego kod odraslih i nema klinički značaj. Ova pojava je zanimljiva pedijatru sa "acetonemskim povraćanjem" povezanim s greškama u prehrani.
Ketonurija u postoperativnom periodu nastaje zbog raspada proteina usled hirurške traume.
U odraslih osoba ketonurija se javlja kod teških oblika dijabetesa i od velike je dijagnostičke vrijednosti. Kod djece može biti sa različitim bolestima, zbog labilnosti metabolizma ugljikohidrata. Stoga čak i manje greške u prehrani, posebno u slučaju akutne infekcije, nervnog uzbuđenja, prekomjernog rada itd. može dovesti do ketoze. Ketonurija se u ranom djetinjstvu može primijetiti s toksikozom, produženim gastrointestinalnim poremećajima, dizenterijom i drugim bolestima. U novorođenčadi je porast ketona u urinu gotovo uvijek uzrokovan neuhranjenošću. Ketonurija, posmatrana kod zaraznih bolesti - škrlatna groznica, gripa, tuberkulozni meningitis i intoksikacija, sekundarni je znak, prolazan je i nema veliku dijagnostičku vrijednost.
Ketonurija za dijabetes razvija se zbog povećane ketogeneze i oslabljene ketolize. Pojačana ketogeneza dovodi do povećane mobilizacije masti iz masnog tkiva, smanjenja stvaranja oksalacetata u Krebsovom ciklusu i smanjenja biosinteze masnih kiselina. Kod teškog dijabetesa, praćenog oštećenjem bubrežnog tkiva (mjesto za raspadanje ketona), nastaje dodatno kršenje ketolize.
Odsustvo glukozurije u prisustvu ketonurije isključuje dijabetes.
Normalno, ketonska tijela nastaju u maloj količini iz konačnog produkta metabolizma ugljikohidrata i lipida - acetil-CoA (C2-telije) kroz acetoacetil-CoA i gotovo u potpunosti se neutrališu.
Kod šećerne bolesti mobilizacija lipida se povećava stvaranjem velike količine acetil CoA (C2telo). istovremeno, uslijed kršenja metabolizma ugljikohidrata, smanjenjem stvaranja oksalacetata, s čime2-tela su uključena u Krebsov ciklus i sagorijevaju se do ugljičnog dioksida i vode. Pored toga, kao rezultat povećanog raspada lipida, reverzna biosinteza C se blokira.2organizam u masne kiseline. Tako se akumulira velika količina C2-body, što dovodi do stvaranja velike količine CoA, a samim tim i acetona, acetooctene i beta-hidroksi-maslačne kiseline, koje se izlučuju mokraćom.
Ketonska tela
Riječ je o srednjim produktima propadanja koji nastaju tokom rada jetre. Obično metabolizam izlaže ketonska tijela daljnjoj razgradnji.
Rezerve glukoze u tijelu nalaze se u tjelesnoj masti, zbog čega je ketonurija jak nedostatak ugljikohidrata. Ketonska tijela su izvrstan dobavljač energije. U tome čak i masne kiseline zaostaju za njima. Stoga, kad mozgu ili srcu nedostaju fosforne kiseline, tijelo odmah stvara ketonska tijela ubrzanim tempom.
Koji su razlozi
Ketonurija je sadržaj ketonskih tijela u urinu iznad normalnog.
U zdravom ljudskom tijelu metabolizam je dobro uravnotežen. Šta može izazvati ketonuriju?

- Preovlađivanje proteina i masti u ishrani. Zbog smanjenja ugljikohidrata u hrani ćelijama nedostaje prehrana. U skladu s tim, razvija se ketonurija. Ovo je reakcija tijela na neravnotežu u prehrani.
- Dijeta i zlouporaba gladi mogu potaknuti pojavu ketonskih tijela u urinu. Pogreške u odabiru prehrane dovode do brzog razgradnje masti, dok se broj enzima povećava. Ketonurija je i pojava acetona u krvi. Obično, ako osoba gladuje duže od šest dana, sadržaj ketonskih tijela u ljudskom tijelu povećava se iznad normalnog.
- Anestezija za operaciju.
- Dijabetes melitus. U ovom se slučaju ketonurija pojavljuje s vremena na vrijeme. Sama ketonska tela nisu uzrok bolesti. Kod dijabetes melitusa propisan je dodatni tijek ugljikohidrata i inzulina.
- Dehidracija. Javlja se kada se tijelo pregrijava ili je komplikacija dijabetesa.
- Bolest jetre ili teške zarazne bolesti (npr. Dizenterija).
- Razvoj tumora u probavnom traktu.
- Poremećaji u gušterači.
- Otrovanje alkoholom ili hemijskim spojevima kao što je fosfor, olovo.
- Oštećenje centralnog nervnog sistema, nervno uzbuđenje.
Sjetite se da miris acetona prilikom disanja ili mokrenja predstavlja signal da se posjetite ljekara. To je i razlog za promjenu načina života i prehrambene ravnoteže.
Pažnja deci
Često se savjetuje s klinikom ako dijete povraća, miris acetona. Ili se takav miris pojavio u urinu. I iako je znak ketonurije, to nije nužno simptom ozbiljne bolesti.

Često se sve objašnjava nesavršenim metaboličkim sistemom. Uzrok ketonurije u djece je stanje u kojem se troši značajna količina energije. Obično se javlja kada:
- emotivni preokret
- povećani fizički napor,
- neuravnotežena prehrana
- prehlada
Činjenica je da djetetovo tijelo nema velike zalihe glikogena, stoga dolazi do aktivnog razgradnje masti i primjećivanja znakova ketonurije.
Trudnoća
Devet meseci trudnoće, žene često moraju da uzimaju testove urina. Ovo je neophodno kako se ne bi promaklo i najmanje odstupanje od norme u tijelu tokom čitavog razdoblja gestacije. Doista, svi organi imaju veliko opterećenje. Što znači da ketonska tela nalaze u urinu?
Obično je njihov mali sadržaj norma. Najjednostavnija analiza bit će prolazak brzog testa. Morate spustiti testnu traku u urinu.
Negativni test smatra se normalnim tokom trudnoće ili je broj ketona minimalan. Ako je test vrijednost od 15 do 160 mg / dl - to izaziva zabrinutost.
Tokom trudnoće ketonurija je alarmantan simptom. Javlja se kod rane toksikoze. Otrovanje tijela, a samim tim i fetusa acetonom, usložnjava tijek trudnoće.

Ako trudnica ima ozbiljan hormonalni neuspjeh, ona prijeti.
Povećavanje nivoa ketona u mokraći novorođenčeta nastaje zbog nedovoljnih grešaka u hranjenju ili prehrani.
Simptomi Ketonurije
Ako je nivo ketona u tijelu značajno porastao, tada će se ketonurija manifestirati:
- iscrpljujuća pospanost
- miris acetona iz usta,
- miris acetona u urinu
- analiza će pokazati visoku razinu bijelih krvnih zrnaca u krvi,
- biohemijski test krvi pokazat će nizak sadržaj glukoze,
Ako je ketonurija izazvala skok acetona u krvi, tada se može pojaviti acetonska kriza.
Značajno povećanje kiselosti u ćelijama može ozbiljno oštetiti unutrašnje organe. U tom slučaju pokreće se zaštitna reakcija - povraćanje.
Simptomi ketonurije u djece:
- pritužbe na bol u trbuhu
- pritužbe na glavobolju
- dijete je umorno, letargično,
- pritužbe na mučninu
- povraćanje
- podizanje temperature na 39 ° C,
- odbijanje hrane
- miriše na aceton
- uvećana jetra
Da li se ketonurija može otkriti?
Samo hemijskim analizama može se otkriti porast razine ketonskih tijela u tijelu. Laboratorija će odmah utvrditi normu ketonskih tijela koja se nalaze u njoj.
U savremenoj medicini, otkriva se ketonurija:
- Lange test,
- Pravni test
- Sample Lestrade,
- modifikovani uzorak Rothere,
- brzi testovi

Brzi testovi su, naravno, najčešći. Njihovo djelovanje temelji se na kemijskoj reakciji čiji je rezultat vidljiv gotovo odmah. Jedino treba staviti test traku u urin ili je staviti na test tabletu. U slučaju pozitivne reakcije, testovi postaju ljubičasti. Svjetlina boje omogućava vam da prosudite po posebnoj ljestvici boja o tome koliko je premašena norma ketonskih tijela.
Međunarodna klasifikacija bolesti
Međunarodna statistička klasifikacija bolesti i povezani zdravstveni problemi (ICD) referentni je vodič kroz koji se materijali mogu usporediti na međunarodnoj razini. Uz njegovu pomoć kombiniraju se različiti metodološki pristupi. Statistika bolesti se nakuplja i klasifikuje. Svakih deset godina IBC pregledava komisija Svjetske zdravstvene organizacije. Da bi se klasificirala i analizirala čitava masa nagomilanih rezultata sa stanovišta statistike, ona se prevodi u alfanumeričke kodove. U takvom se radu koristi razvijeni ICD. Danas to odgovara ICD-10. Sadrži 22 sata (odjeljenja).
Prema ICD direktorijumu, ketonuria ima oznaku R82.4.
Prevencija i dijeta ketonurije
Za prevenciju ketonurije potrebno je:
- jedi pravo
- vodite zdrav način života:
- što je više moguće na svježem zraku,
- umerena vežba
- Nemojte započeti hronične bolesti.

Kod hronične bolesti poput dijabetesa ljekar se sistematski savjetuje. Povremeno je potrebno napraviti ekspresni test.
Pozitivna reakcija ukazuje na patologiju u tijelu. Obratite pažnju na signal! Hitna žalba na kliniku će brzo vratiti tijelo u normalu. Bez odstupanja od prehrane koju je propisao ljekar, možete ubrzati oporavak.
Ketonuria dijeta:
- nemojte jesti hranu koja sadrži visoku količinu proteina i masti,
- redovan unos ugljenih hidrata
- pijte više vode i sode otopine (kod dijabetesa - inzulina).
Jelovnik treba da uključuje: kuvanu govedinu i zec, razne juhe od povrća, ribu s malo masti. Korisne žitarice bez maslaca, povrća i voća. Preporučuje se piti više sokova, voćnih napitaka, kompota.
Isključi iz menija:
- masno meso
- začinjene začine
- slatko
- agrumi
- banane
- gljive
- brza hrana.
Fokusirajte se na lečenje
Ketonurija se ne smije tretirati kao odvojena bolest. Ona je samo posljedica. Treba isključiti uzroke koji su ga uzrokovali. Prvo je potreban potpuni pregled. Točnost dijagnoze i utvrđivanje uzroka ketonurije jamče uspješno liječenje.
Ljekari daju nekoliko savjeta:
- Ako imate prekomjernu težinu, povremeno bi trebali organizirati dane posta.
- Ako je analiza otkrila porast ketona u urinu, kupite testove kako biste ih koristili kod kuće.

- Urin treba prikupiti na analizu najkasnije četiri sata prije porođaja.
- Dijete se može piti alkalnim pićem svakih 10-15 minuta u malim porcijama. Aktivni ugalj, enterosgel će pomoći da se očisti probavni trakt.
- S nadolazećim povraćanjem korisno je piti frakcijski napitak. Pozovite hitnu pomoć.
Kako liječiti ketonuriju, postoje savjeti u narodnoj medicini.
- Uz učestalo povraćanje, pijte mineralnu vodu, kompot od sušenog voća, otopinu glukoze. Jedna kašika u deset minuta.
- Stavite klistir kod kuće. Prvo voda na sobnoj temperaturi, a kasnije topla, u koju se dodaje žličica sode.
- Popijte piće: otopite 2 kašike u 1 litri vode. med, sipaj sok jednog limuna. Pijte 1 kašiku. svakih 15 minuta.
- Recept za otopinu sode: otopite 1 žličicu sode u 250 ml vode. Pijte 1 kašika kašike. svakih 10 minuta.
- Uzmite decokcije umirujućeg bilja.
- Da biste ubrzali izbacivanje toksina iz organizma, jedite malo po malo. Bolje je jesti samo krekere.
Liječenje ovisi o individualnim karakteristikama tijela. Morate ga strogo kontrolirati lekar.
76. Holesterol. Načini unosa, upotrebe i izlučivanja iz organizma. Serumski holesterol. Biosinteza holesterola, njegove faze. Regulacija sinteze
Holesterol je steroid specifičan za životinjske organizme. Sintetizira se u mnogim ljudskim tkivima, ali glavno mjesto sinteze je jetra. U jetri se sintetiše više od 50% holesterola, u tankom crijevu - 15-20%, ostatak holesterola sintetiše se u koži, nadbubrežnoj kore i žlijezdama. Oko 1 g holesterola se sintetiše dnevno u tijelu, 300-500 mg se unosi s hranom (sl. 8-65). Holesterol obavlja brojne funkcije: dio je svih staničnih membrana i utječe na njihova svojstva, služi kao početni supstrat u sintezi žučnih kiselina i steroidnih hormona. Prekursori u metaboličkom putu sinteze holesterola pretvaraju se i ubikinon, komponentu respiratornog lanca i dolichol, koji je uključen u sintezu glikoproteina. Zbog svoje hidroksilne skupine, holesterol može tvoriti estere s masnim kiselinama. Eterificirani holesterol prevladava u krvi i pohranjuje se u malim količinama u nekim vrstama ćelija koje ga koriste kao supstrat za sintezu drugih tvari. Holesterol i njegovi esteri su hidrofobni molekuli, pa se krvlju prenose samo kao dio različitih vrsta lijekova. Razmjena kolesterola je izuzetno složena - samo za njenu sintezu potrebno je oko 100 uzastopnih reakcija. Ukupno, oko 300 različitih proteina je uključeno u metabolizam holesterola. Poremećaji metabolizma holesterola dovode do jedne od najčešćih bolesti - ateroskleroze. Smrtnost od posljedica ateroskleroze (infarkt miokarda, moždani udar) vodi u ukupnoj strukturi smrtnosti. Ateroskleroza je "poligenska bolest", tj. u njegovom je razvoju uključeno mnogo faktora od kojih su najvažniji nasljedni. Akumulacija holesterola u tijelu dovodi do razvoja još jedne uobičajene bolesti - žučne kamenačke bolesti.
A. Sinteza holesterola i njegova regulacija
Reakcije sinteze holesterola nastaju u citosolu ćelija. Ovo je jedan od najdužih metaboličkih puteva u ljudskom tijelu.
Fenilketonurija

Fenilketonurija - nasljedno kršenje metabolizma aminokiselina zbog nedostatka jetrenih enzima koji su uključeni u metabolizam fenilalanina u tirozin. Rani znakovi fenilketonurije su povraćanje, letargija ili hiperaktivnost, miris plijesni iz urina i kože, odložen psihomotorni razvoj, tipični kasni znakovi uključuju oligofreniju, kašnjenje u fizičkom razvoju, konvulzije, ekzematozne promjene na koži itd. Screening novorođenčadi na fenilketonuriju provodi se čak i u rodilištu, naknadna dijagnostika uključuje molekularno genetičko testiranje, određivanje koncentracije fenilalanina u krvi, biohemijsku analizu urina, EEG i MRI mozga. Liječenje fenilketonurije je pridržavanje posebne prehrane.

Opće informacije
Fenilketonurija (Fellingova bolest, fenilpiruvična oligofrenija) je urođena, genetski određena patologija koju karakterizira poremećena hidroksilacija fenilalanina, nakupljanje aminokiselina i njegovih metabolita u fiziološkoj tekućini i tkivima, praćeno ozbiljnim oštećenjima središnjeg živčanog sustava. Fenilketonuriju je prvi put opisao A. Felling 1934. godine, javlja se s učestalošću od 1 slučaja na 10 000 novorođenčadi. U neonatalnom razdoblju fenilketonurija nema kliničke manifestacije, međutim unos fenilalanina s hranom uzrokuje manifestaciju bolesti već u prvoj polovici života, a posljedično dovodi do teških poremećaja u razvoju djeteta. Zato je pre-simptomatsko otkrivanje fenilketonurije u novorođenčadi najvažniji zadatak neonatologije, pedijatrije i genetike.
Uzroci fenilketonurije
Fenilketonurija je autosomno recesivni poremećaj nasljeđivanja. To znači da dijete za razvoj kliničkih znakova fenilketonurije mora naslijediti jednu neispravnu kopiju gena od oba roditelja, koji su heterozigotni nosači mutirajućeg gena.
Najčešće, razvoj fenilketonurije prouzrokovan je mutacijom u genu koji kodira enzim fenilalanin-4-hidroksilaza smješten na dugom kraku kromosoma 12 (lokus 12q22-q24.1). To je takozvana klasična fenilketonurija tipa I, koja čini 98% svih slučajeva bolesti. Hiperfenilalaninemija može doseći 30 mg i više. Ako se ne liječi, ovu varijantu fenilketonurije prati duboka mentalna retardacija.
Pored klasičnog oblika, razlikuje se atipična fenilketonurija koja nastavlja iste kliničke simptome, ali nije podložna korekciji dijetalnom terapijom. Tu spadaju fenilketonurija tipa II (nedostatak dehidroterterin reduktaze), fenilketonurija tip III (deficit tetrahidrobiopterina) i druge, retke varijante.
Vjerojatnost rođenja djeteta sa fenilketonurijom povećava se s bliskim brakovima.
Patogeneza fenilketonurije
Klasični oblik fenilketonurije zasnovan je na insuficijenciji enzima fenilalanin-4-hidroksilaze koji sudjeluje u pretvorbi fenilalanina u tirozin u mitohondriju hepatocita. Zauzvrat, tirozin derivat tirozina polazni je materijal za sintezu kateholamina (adrenalin i norepinefrin), a diiodotirosina za stvaranje tiroksina. Pored toga, stvaranje melaninskog pigmenta rezultat je metabolizma fenilalanina.
Nasljedni nedostatak enzima fenilalayin-4-hidroksilaza u fenilketonuriji dovodi do kršenja oksidacije fenilalanina iz hrane, što rezultira njegovom koncentracijom u krvi (fenilalaninemija) i cerebrospinalnom tečnošću, a nivo tirozina u skladu s tim opada. Višak fenilalanina eliminira se povećanim izlučivanjem urina njegovih metabolita - fenilpirovične kiseline, fenilmilaktične i feniloctene kiseline.
Prekid metabolizma aminokiselina prati oštećena mijelinizacija živčanih vlakana, smanjenje stvaranja neurotransmitera (dopamin, serotonin itd.), Što pokreće patogenetičke mehanizme mentalne retardacije i progresivnu demenciju.
Simptomi fenilketonurije
Novorođenčad s fenilketonurijom nema kliničke znakove bolesti. Tipično, manifestacija fenilketonurije kod dece javlja se u dobi od 2-6 meseci. S početkom hranjenja, protein majčinog mlijeka ili njegovi nadomjesci počinju ući u bebino tijelo što dovodi do razvoja prvih, nespecifičnih simptoma - letargije, ponekad anksioznosti i hiperpobuđenosti, regurgitacije, mišićne distonije, konvulzivnog sindroma. Jedan od ranih patognomoloških znakova fenilketonurije je uporno povraćanje, što se često pogrešno smatra manifestacijom pilorične stenoze.
Do druge polovine godine detetov zaostajanje u psihomotornom razvoju postaje uočljiv. Dijete postaje manje aktivno, ravnodušno, prestaje prepoznavati voljene osobe, ne pokušava sjediti i stati na noge. Nenormalan sastav urina i znoja izaziva karakterističan „mišji“ miris (miris plijesni) koji proizlazi iz tijela. Često dolazi do ljuštenja kože, dermatitisa, ekcema, skleroderme.
U djece sa fenilketonurijom koja ne primaju liječenje, otkriva se mikrocefalija, prognatija, kasnije (nakon 1,5 godine) zuba, hipoplazija cakline. Primjećuje se kašnjenje u razvoju govora i otkrivaju se 3-4 godine duboka oligofrenija (idiotizam) i gotovo potpuno odsutnost govora.
Djeca s fenilketonurijom imaju displastičnu tjelesnost, često prirođene srčane mane, autonomne disfunkcije (znojenje, akrocijanoza, arterijska hipotenzija) i pate od opstipacije. Fenotipske karakteristike dece koja pate od fenilketonurije uključuju svetlu kožu, oči i kosu. Dete sa fenilketonurijom odlikuje se specifičnim položajem „krojača“ (gornji i donji udovi savijeni u zglobovima), drhtanje ruku, kolebanje, potez od mlevanja i hiperkineza.
Kliničke manifestacije tipa II fenilketonuriju karakteriše jak stepen mentalne retardacije, povećana razdražljivost, napadaji, spastička tetrapareza i hiperrefleksija tetiva. Napredovanje bolesti može dovesti do smrti djeteta starosti od 2 do 3 godine.
Kada fenilketonuri tipa III razvije trijadu znakova: mikrocefalija, oligofrenija, spastična tetrapareza.
Dijagnoza fenilketonurije
Trenutno je dijagnoza fenilketonurije (kao i galaktozememije, urođena hipotireoza, adrenogenitalni sindrom i cistična fibroza) dio neonatalnog programa probira za novorođenčad.
Screening test se provodi na 3-5 dana života punog i 7 dana života prevremeno rođene djece uzimanjem uzorka kapilarne krvi na posebnom papirnom obliku. Ako se otkrije hiperfenilalanemija, više od 2,2 mg djeteta upućuje se na pedijatrijsku genetiku radi ponovnog pregleda.
Da bi se potvrdila dijagnoza fenilketonurija, provjerava se koncentracija fenilalanina i tirozina u krvi, određuje se aktivnost jetrenih enzima (fenilalanin hidroksilaza), obavlja se biohemijski pregled urina (određivanje ketonske kiseline), metaboliti kateholamina u urinu i dr. Provodi se EEG-om i MRI mozga, a dijalogom djeteta se dodatno pretražuje.
Genetska oštećenja fenilketonurije mogu se otkriti čak i tijekom trudnoće tijekom invazivne prenatalne dijagnoze fetusa (horionbiopsija, amniocenteza, kordocenteza).
Diferencijalna dijagnoza fenilketonurije provodi se s intrakranijalnom rođenom traumom kod novorođenčadi, intrauterinim infekcijama i drugim metaboličkim poremećajima aminokiselina.
Liječenje fenilketonurijom
Temeljni faktor u lečenju fenilketonurije je dijeta koja ograničava unos proteina u organizam. Liječenje se preporučuje započeti koncentracijom fenilalanina> 6 mg%. Razvijene su posebne mješavine za novorođenčad - Afenilak, Lofenilak, za djecu stariju od 1 godine - Tetrafen, Fenil, stariji od 8 godina - Maxamum-XP i dr. Osnova prehrane je hrana bez proteina - voće, povrće, sokovi, proteinski hidrolizati i smjese aminokiselina. . Proširenje prehrane moguće je nakon 18 godina u vezi s povećanjem tolerancije na fenilalanin. U skladu s ruskim zakonom, pružanje medicinske ishrane ljudima koji pate od fenilketonurije trebalo bi biti besplatno.
Pacijentima se propisuje unos mineralnih jedinjenja, vitamina grupe B itd., Prema indikacijama - nootropni lijekovi, antikonvulzivi. U složenoj terapiji fenilketonurije naširoko se koriste opšta masaža, terapija vežbanjem i akupunktura.
Djeca koja pate od fenilketonurije nalaze se pod nadzorom lokalnog pedijatra i neuropsihijatra, a često im je potrebna pomoć logopeda i patologa. Potrebno je pažljivo praćenje neuropsihološkog statusa djece, praćenje nivoa fenilalanina u krvi i pokazatelja elektroencefalograma u krvi.
Atipični oblici fenilketonurije koji se ne mogu liječiti dijetom zahtijevaju imenovanje hepatoprotektora, antikonvulziva, nadomjesne terapije levodopom, 5-hidroksitriptofanom.
Predviđanje i prevencija fenilketonurije
Provođenje masovnog probira na fenilketonuriju u neonatalnom razdoblju omogućuje vam da organizirate ranu dijetalnu terapiju i spriječite teška oštećenja mozga, oslabljenu funkciju jetre. Uz rano imenovanje eliminacijske dijete za klasičnu fenilketonuriju, prognoza za razvoj dece je dobra. S kasnim liječenjem, prognoza za mentalni razvoj je loša.
Prevencija komplikacija fenilketonurije sastoji se u masovnom pregledu novorođenčadi, ranom propisivanju i dugoročnom poštivanju dijetalne prehrane.
Da bi se procijenio rizik rođenja djeteta s fenilketonurijom, potrebno je dati preliminarno genetsko savjetovanje parovima koji već imaju bolesno dijete, u srodnoj su vezi i imaju rođake s ovom bolešću. Žene s fenilketonurijom koje planiraju trudnoću trebale bi pridržavati stroge prehrane prije začeća i tijekom trudnoće kako bi se isključilo povećanje razine fenilalanina i njegovih metabolita i oštećen razvoj genetski zdravog fetusa. Rizik da rodi dijete sa fenilketonurijom kod roditelja koji imaju defektni gen je 1: 4.
Pogledajte video: Natrijum u krvi Na (Novembar 2024).



-
Hladna supa
Kolekcije recepata Recepti hladnih supa Recepti pronađeni sa fotografijama - 111 kom. Kuhana repa - 1 kom. Kuvana jaja - 3 kom. Peršun - 0,5 hrpa kopar - 0,5 hrpa luk zeleni - 3 kom. Morska sol - po ukusu Paprika ch.m. ... -

-

-